BC/NW 2008, №1 (12):16.3
ПРОБЛЕМЫ ПАРАЛЛЕЛЬНОЙ
РЕАЛИЗАЦИИ АЛГОРИТМОВ РАСЧЕТА ЭЛЕКТРОННОЙ СТРУКТУРЫ БОЛЬШИХ МОЛЕКУЛ НА
КЛАСТЕРНЫХ СИСТЕМАХ
А.М. Чернецов, О.Ю.Шамаева О.Ю.
(Москва,
Московский Энергетический Институт (ТУ), Россия)
1. Введение
Расчеты электронной структуры
гигантских молекул являются одними из самых сложных в современной науке и
требуют использования высокопроизводительных вычислительных средств, таких как
суперЭВМ и кластерные установки. Расчеты электронной структуры, в частности,
биомолекул (белков, ДНК) и наночастиц, актуальны для ряда областей науки:
химии, биохимии, физики конденсированного состояния вещества и др. В
практическом плане эти расчеты важны для фармакологии, нанотехнологий,
исследований явлений сверхпроводимости, разработки квантовых компьютеров и др.
Существуют строгие и
полуэмпирические методы вычисления для решения задачи. Строгие методы имеют
асимптотическую сложность O (N5) и выше, где N – размерность задачи,
определяемая количеством и типом атомов (орбиталей). Даже для вычисления
небольших молекул порядка 2000 атомов необходимо больше чем 1 месяца для
полного расчета молекулы.
Поэтому для больших
молекулярных систем целесообразно использовать методы нулевого
дифференциального перекрывания (НДП) квантовой химии, которые имеют сложность O
(N3). Эта сложность определяется матричной диагонализацией (решение
симметричной задачи собственных значений) и неприемлема для больших N, даже при
использовании супер-ЭВМ.
Чтобы вычислять
квантово-химические свойства, необходима
матрица плотности P, являющаяся
функцией собственных векторов. Недавно были разработаны современные модификации
полуэмпирических методов вычисления, имеющего сложность O (N) для разреженных
матриц. Общая схема существующих методов решения задачи расчета электронной
структуры молекул представлена на рис. 1.
Существующие программы расчета
электронной структуры, включая дорогостоящие коммерческие, или не имеют
параллельной версии вообще, или плохо распараллеливаются. Поэтому реализация
таких расчетов требует больших временных затрат и ресурсов памяти.
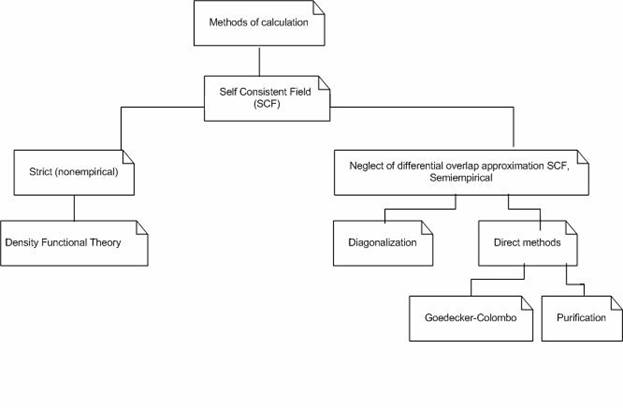
Рис. 1. Общая схема основных методов расчета
электронной структуры молекул.
2. Строгие методы расчета
электронной структуры
Строгие не эмпирические методы
квантовой химии (квантовой механики молекул), учитывающие электронную
корреляцию, имеют асимптотическую сложность O (N5) и выше, которая
для вычислений больших молекулярных систем является абсолютно недопустимой.
Например, полное вычисление молекулы только из 1738 атомов требует время
больше, чем один год.
Рассмотрим одноэлектронные
методы расчета. В них для определения состояния многоэлектронной системы,
имеющей минимальный уровень полной энергии, используется концепция эффективного
одноэлектронного гамильтониана, который представляется в виде:
![]() (1)
(1)
Здесь ![]() – константа Планка, me – масса электрона, v –
оператор потенциальной энергии,
– константа Планка, me – масса электрона, v –
оператор потенциальной энергии, ![]() – оператор Лапласа, r – радиус-вектор. В квантовой механике
молекул и квантовой химии принято использовать так называемую атомную систему
единиц, в которой константа Планка и масса электрона приняты за единицу [1]. В
дальнейшем изложении предполагается
использование атомной системы единиц. При этом возникает задача на
собственные значения
– оператор Лапласа, r – радиус-вектор. В квантовой механике
молекул и квантовой химии принято использовать так называемую атомную систему
единиц, в которой константа Планка и масса электрона приняты за единицу [1]. В
дальнейшем изложении предполагается
использование атомной системы единиц. При этом возникает задача на
собственные значения
![]() (2),
(2),
где i нумерует собственные значения
и соответствующие собственные вектора. Нормализованные собственные вектора
этого оператора (одноэлектронные волновые функции yi) описывают одноэлектронную
плотность
![]() (3),
(3),
где ni – “заселенность” i-го
состояния, равная 0 или 1. Электроны занимают все нижние уровни энергии, т.е. ![]() , где ef отвечает высшему заселенному
уровню. Здесь q(x) -
ступенчатая функция Хевисайда. Заселенности могут рассматриваться как
собственные значения более общего оператора, характеризуемого матрицей
плотности
, где ef отвечает высшему заселенному
уровню. Здесь q(x) -
ступенчатая функция Хевисайда. Заселенности могут рассматриваться как
собственные значения более общего оператора, характеризуемого матрицей
плотности
![]() (4),
(4),
которая является идемпотентной, т.е.
![]() (5)
(5)
Энергия основного состояния
системы, как и другие физико-химические свойства молекулярной системы, могут
быть выражены через матрицу плотности [1].
Строгая схема основывается на
методе Хартри-Фока, в котором в качестве эффективного одноэлектронного
гамильтониана применяется так называемый оператор Фока. Кроме того, применяются
различные схемы метода функционала плотности (DFT), также относящегося к одноэлектронным
методам. Эти схемы относятся к группе методов самосогласованного поля (ССП)
[1]. Эффективные одноэлектронные гамильтонианы в этих методах зависят от
матрицы плотности Р.
В методах ССП сначала
рассчитывается начальное приближение к Р
, затем из Р рассчитывается
одноэлектронный гамильтониан, с ним решается задача на собственные значения, из
собственных векторов рассчитывается новая Р, из нее строится новый гамильтониан, затем процедура итерационно
повторяется. На рис.2 приведена общая схема процедуры расчетов по методу
самосогласованного поля.
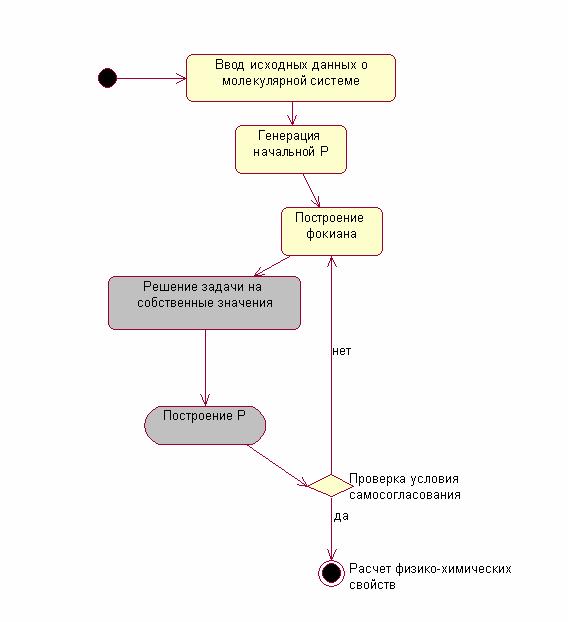
Рис.2. Общая процедура расчетов
методом ССП.
Серым цветом отмечены блоки,
определяющие общее время расчета. Именно эти блоки подлежат параллельной
модификации с целью сокращения времени решения.
Уравнения метода ССП (см. (2))
фактически являются системой интегро-дифференциальных уравнений, для решения
которых применяется метод Ритца, когда одноэлектронные функции - молекулярные
орбитали (МО) yi разлагаются в конечный ряд
(являются линейной комбинацией) по заданным базисным функциям (атомным
орбиталям, АО), что дало название методу – ССП МО ЛКАО (самосогласованного поля
молекулярных орбиталей линейной комбинации атомных орбиталей ).
![]() (6)
(6)
Тогда задача фактически
сводится к нахождению коэффициентов разложения молекулярных орбиталей по
атомным орбиталям. Асимптотическая сложность строгого неэмпирического метода
ССП МО ЛКАО – O(N4), где N – размерность базиса, что также является
абсолютно неприемлемым с точки зрения расчета для интересующих нас сверхбольших
молекулярных систем. Существуют специализированные расчетные схемы, имеющие
меньшую асимптотическую сложность, однако они также недостаточно эффективны для
задач больших размерностей.
Строгие неэмпирические
вычисления электронной структуры больших белков, ДНК и наноструктур фактически
не могут быть реализованы на существующих средствах вычислительной техники. Мы
иллюстрируем это на следующем примере. Вычисление молекулы даже такого
маленького белка, как цитохром-C (1738
атомов) с использованием метода DFT (один
из вариантов метода ССП) по специализированной программе ProteinDFT на
кластере, содержащем 15 микропроцессоров Hewlett-Packard Alpha 21264, занимает больше чем один день для
одной итерации ССП. Полное вычисление занимает больше одного месяца, даже без
оптимизации геометрии. Трудоемкость расчета больших молекулярных систем,
насчитывающих более 103 атомов, традиционными методами не позволяет
осуществлять вычисления за приемлемое время.
3. Упрощенные полуэмпирические
методы нулевого дифференциального перекрывания
Рассмотренная выше строгая
схема метода ССП МО ЛКАО является
основой для построения полуэмпирических
моделей в приближении нулевого дифференциального перекрывания (НДП) [1,7], в
которых резко упрощается расчет матрицы оператора Фока в базисе атомных
орбиталей (стадии расчета, определяющей основное время вычислений в неэмпирическом методе ССП МО ЛКАО). В этом
случае базис АО предполагается ортонормированным, и уравнения ССП МО ЛКАО
сводятся к задаче на собственные значения с симметричной матрицей фокиана:
Hci = Eici (7),
где H –
симметричная матрица Фока (фокиан), сi и Еi
–
i-е
собственные вектора и собственные значения
матрицы H.
Собственные вектора сi ,
представляемые вектором-столбцом, являются ортонормированными. Составим из
векторов-столбцов сi матрицу С:
C = (c1 | c2| … |cN) (8),
где N – размерность базиса. Тогда
CTC = IN (9),
где IN –
единичная матрица размерности N, а
верхний индекс T у
матрицы означает ее транспонирование.
Матрица плотности Р в базисе АО рассчитывается из
матрицы собственных векторов С
как
P= CnCT (10),
где n-
диагональная матрица, содержащая на диагонали величины заселенностей (см.
(.3)). Иначе говоря, n является диагонально-матричным
представлением ступенчатой функции Хевисайда q(x).
Симметричная матрица P является идемпотентной, т.е.
P2 = P (11)
и коммутирует с фокианом:
PH=HP (12).
Для определения матрицы
плотности P,
в соответствии с (3), предполагается, что электроны заселяют все низшие
одноэлектронные уровни энергии Ei вплоть до максимального
значения (энергии Ферми ef), а все более высокие уровни
энергии остаются незанятыми. Т.к. общее число занятых одноэлектронных уровней
равно половине числа электронов Ne в молекулярной системе, то соответствующее условие
tr
P = Ne/2 (13).
Вместо прямого решения задачи
на собственные значения (7) с последующим нахождением матрицы плотности P по
формуле (10), теоретически возможно нахождение симметричной коммутирующей с H
идемпотентной матрицы P, удовлетворяющей условиям (11-13), что при
условии заполнения низших одноэлектронных уровней энергии является
эквивалентной формулировкой.
Основное время при решении
задачи на собственные значения (7) расходуется уже не на построение матрицы
Фока (фокиана), как это было в строгом неэмпирическом методе, а на
диагонализацию (нахождение собственных векторов и собственных значений) этой
матрицы.
При этом в соответствии со
схемой на рис. 2 диагонализация итерационно повторяется. В реальной расчетной
практике требуемое количество диагонализаций матриц оказывается еще во много
раз больше. Дело в том, что общая схема метода ССП на рис. 2 предполагает
проведение расчета для фиксированной геометрии молекулы, когда декартовы
координаты ее ядер зафиксированы.
На самом деле необходима
оптимизация геометрии, т.е. нахождение минимума расчетной полной энергии
молекулярной системы по отношению к декартовым координатам ядер. В этом случае
вся описанная итерационная расчетная схема является частью более общей процедуры, в процессе
которой осуществляется минимизация полной энергии как нелинейной функции многих
переменных (координат ядер) с применением традиционных численных методов.
Например, методы, использующие аналитические производные полной энергии по
декартовым координатам ядер, в частности, методы типа сопряженных градиентов.
Тогда полное число требуемых диагонализаций фокиана, можно оценить как
произведение числа итераций при фиксированной геометрии на число шагов
процедуры оптимизации геометрии. Требуемое число итераций при фиксированной
геометрии не является постоянной величиной.
Конечно, точность расчетов
полуэмпирическими методами НДП резко уступает неэмпирическим методам, но такие
полуэмпирические схемы могут использоваться и для первоначальных оценок, и для
оценок на качественном уровне, не требующих результатов высокой количественной
точности [1].
Имеются различные варианты
полуэмпирических схем НДП, которые практически не отличаются с точки зрения
времени расчета больших молекулярных систем. Их отличия определяются
параметризацией, задающей формулы для расчета фокиана. Это дает возможность
достигать различного уровня точности расчета, но никак не меняет основной
численной процедуры (в традиционном подходе – диагонализации матриц, см. ниже),
для которой фокиан является просто входным параметром. В данной работе была
использована одна из современных параметризаций – метод AM/1.
4. Оценки компьютерных ресурсов
для диагонализации симметричных матриц
Проблемы расчета электронной
структуры сверхбольших молекулярных систем не ограничиваются сверхвысокими
требованиями к процессорному времени расчета. Подобные расчеты невозможными не
только на самых мощных современных суперЭВМ, но даже при их объединении в GRID-инфраструктуру. Другая проблема связана с
требованиями к емкости оперативной памяти, которые также быстро возрастают с
ростом N до
неприемлемо высоких значений – десятков гигабайт.
Среди методов расчета
электронной структуры, требующих больших объемов оперативной памяти, можно
указать на эффективные с точки зрения производительности "in-core"
(работа исключительно в оперативной памяти) методы неэмпирической квантовой
химии, реализованные, например, в известных комплексах программ серии Gaussian. Так, для “in-core”
методов ССП и MP2
требуется оперативная память емкостью O(N4), где N - размерность базиса. Для “in-core”
метода ССП при N=200
требуется 1.6 Гбайт оперативной памяти, а для N=300 - уже 8.1 Гбайт. Однако у этих методов
имеются и альтернативные численные схемы, требующие оперативной памяти меньшей
емкости, например, O(N3) для прямого метода MP2 (включая расчет градиентов полной
энергии). Большинство типичных методов расчета по этим программам требует
объемов оперативной памяти порядка O(N2).
Для более крупных молекул или
более точных методов расчета (MP4, CCSD, аналитические вторые производные энергии
метода MP2 и
др.) требуется существенно большая емкость оперативной памяти.
При больших N применение диагонализации становится
практически неприемлемым, когда даже для 64-разрядных систем, т.е. при
отсутствии формальных ограничений в 2-3 Гбайт на емкость памяти в 32-разрядных
системах х86-архитектуры.
Однако большинство интересующих
нас сверхбольших молекулярных систем, в силу их физико-химической природы, при
расчете характеризуются разреженными матрицами фокиана. В результате в
последние годы появились специализированные численные методы расчета,
обладающие (при использовании технологии разреженных матриц) линейным
масштабированием процессорного времени O(N),
которые могут быть использованы в полуэмпирических схемах НДП. В методах
расчета с учетом разреженности требования к оперативной памяти также растут
линейно.
5. Современная числовая альтернатива
моделей к диагонализации матриц: очистка
Существуют некоторые
современные числовые модели [2,3], из которых мы рассмотрим модель “очистки”
[3,4].
Общая идея использования
очистки (purification) для построения
матрицы плотности основывается на итерационном разложении гамильтониана по
непрерывно возрастающему полиному P(x),
x Î [0,1]
с зафиксированным экстремумом между 0 и 1, т.е. P(0)=0, P(1)=1 и P¢(0)= P¢(1)=0.
Можно показать [4], что вложенная последовательность таких многочленов сходится
к ступенчатой функции со ступенькой где-то на [0,1].
Метод классической “очистки”
McWeeny [3] состоит в проведении итерационного процесса по формуле
![]() (14),
(14),
где P -
матрица плотности. Процесс останавливается при достижении заданной точности по
идемпотентности, т.е.
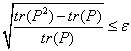 (15).
(15).
В результате ряда исследований
были найдены более быстро сходящиеся методы, являющиеся модификациями метода
классической “очистки”. Одним из них является модификация
Пальцера-Манолополиса, называемая также канонической очисткой [4]. В этом
методе итерационная формула для нахождения матрицы плотности P выглядит следующим образом:
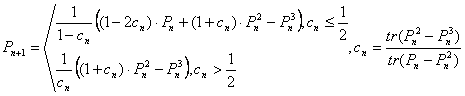 , (16),
, (16),
здесь n – номер шага
итерационного процесса, tr(P) – след
матрицы P.
Выбор начального значения P0 рассмотрен в [4,5].
6. Использование свойств
разреженности матрицы плотности
В общем случае матрица
плотности имеет сложную структуру. Для расчетов целесообразно использовать
гиперматричную схему её хранения в памяти [6].
Рассмотрим особенности разработки
параллельного алгоритма вычисления матрицы плотности методом
Пальцера-Манополиса для разреженных матриц с блочно – трехдиагональным
портретом [7]. Пример такой матрицы для случая 3 квадратных блоков размерностей
m1, m2 и m3 соответственно, приведен на рис. 3. Количество блоков ограничено
доступными ресурсами.
Такую матричную структуру имеют
некоторые типы органических молекул, например полимеры, линейные несвёрнутые
протеины и большие фрагменты линейных цепей ДНК/РНК.
Для рассматриваемого типа разреженности
и нашей задачи необходимо реализовать алгоритмы линейных операций и матричного
умножения.
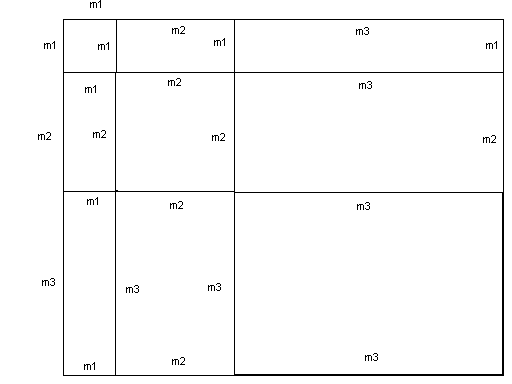
Рис. 3. Блочно трехдиагональная матрица.
При умножении
блочно-трехдиагональных матрицах в матрице результата появляется одна блочная
диагональ, отличная от нуля. Однако, из свойств квантовой химии известно, что
при таком расширении элементы отличные от нуля, гасятся, и мы можем исследовать
только умножение трех диагоналей блоков.
На рис. Приведены исходные А и
В и результирующая R матрицы, для которых ниже приведены основные расчетные
формулы.
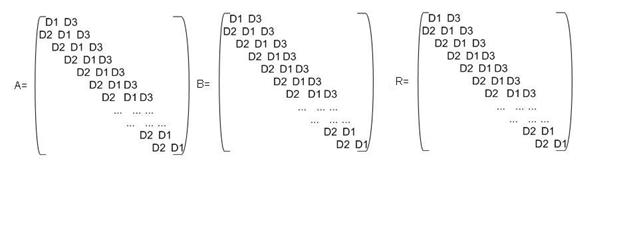
Это необходимо, чтобы
разработать специальную схему хранения блочно-трехдиагональных матриц.
Предлагается хранить матрицу как три 3-мерных массива D1, D2 и D3. Первые два
индекса соответствуют блоку. Последний индекс соответствует позиции блока в
соответствующей диагонали.
В блочном представлении матрица
выглядит так:
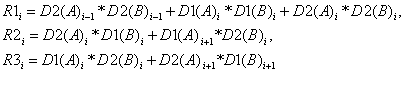 (17)
(17)
Здесь
D1 – матрица блоков главной диагонали, D2 и D3 – соответственно матрицы блоков
нижней поддиагонали и верхней наддиагонали, R1, R2 и R3 - соответствующие
результирующие матрицы блоков.
Если
учесть, что матрица Н является симметричной, а блоки главной диагонали –
квадраты - то становится возможным
сэкономить память на хранении массива D3 и после упрощений получим:
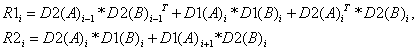 (18)
(18)
Случаи для крайних блоков
(первого и последнего) легко выводятся из общих формул и поэтому здесь опущены.
В описанном случае блочно
трехдиагональных матриц необходимо хранить одновременно 4 разреженных матрицы:
3 для P, P2 и P3 и еще одна матрица необходима для
хранения матрицы фокиана. Требуемая память может быть рассчитана как
![]() байт,
байт,
где dim – максимальная
размерность мелких блоков, nbl –
число блоков в главной диагонали. Следовательно для 2 Гбайт памяти и
размерности, равной 250, мы можем хранить фокиан с максимальной
размерностью150000 AO
(nbl=750). В то же самое время для метода Пальцера-Манолополиса в плотной форме
максимальная размерность матриц составляет 8500 AO.
7. Параллельные решения для MPI кластеров
Анализ приведенных выше
матричных расчетных формул (16-18) позволяет разработать
параллельно-последовательную схему вычислений, приведенную на рис. 4. Данная
схема содержит итерационно повторяемую параллельно-последовательную часть,
число повторений которой зависит от выполнения условия (15).
Приведенная схема положена в
основу параллельной программы расчета, которая была реализована на языке Fortran 95 с использованием
инструментальных средств MPI.
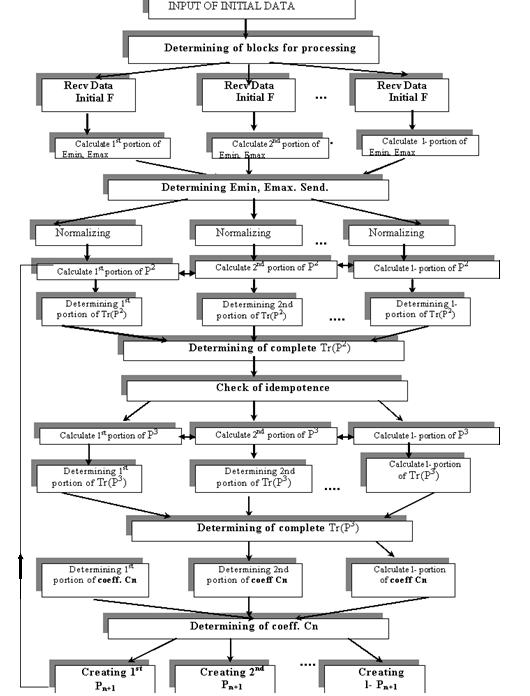
Рис. 4. Общая схема
параллельной модификации метода Пальцера-Манолополиса.
При тестировании программы при
вычислении молекулы полипептида с 3000 орбиталей (около 1000 атомов) были
получены идентичные матрицы плотности, как для разреженного варианта алгоритма
решения, так для плотного варианта, что подтвердило обоснованность и
корректность использования разреженности.
При использовании кластера,
основанного на МП Intel Xeon
Pentium 4/2.6 Ghz и
межсоединении Myrinet 2000
(пиковая пропускная способность - 250 Mbytes/s, mpich-gm 1.2.6-14b, gm
v. 2.0.2), были получены
результаты, представленные на рис. 5.
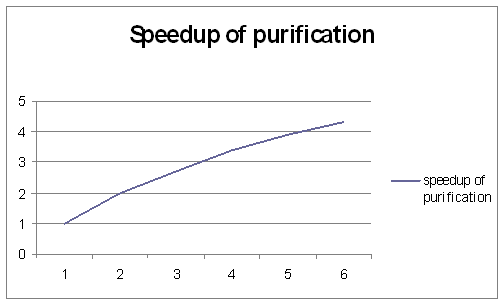
Рис. 4. Ускорения, достигнутые
методом Пальцера-Манолополиса.
Можно сравнить достигнутые
ускорения для кластера, основанного на Intel Xeon с результатами лучшей коммерческой
программы MOPAC2002 [8] для расчета молекулы фуллерена C960 на SMP системе Origin300. Полученные значения
ускорения ниже, чем в нашей программе с числом процессоров до 6.
8. Заключение.
В перспективе предполагается
расширить исследуемый класс разреженных матриц плотности и разработать соответствующие
эффективные алгоритмы их вычисления.
Авторы выражают их
благодарность ВЦ РАН за доступ к его кластерным ресурсам для отладки и
тестирования программы.
Работа выполнена при частичной
финансовой поддержке РФФИ, проект N 05-07-08031-офи-a.
9.
Литература.
[1] Н.Ф. Степанов, “Квантовая механика и квантовая химия”,
Изд-ва “Мир” и “МГУ”, М., 2001, 518 с.
[2] Roi Baer, Martin
Head-Gordon “Chebyshev expansion methods for
electronic structure calculations on large molecular systems”, J. Chem. Phys.,
107(28),
[3] McWeeny R., Rev. Mod.
Phys. 32, 1960, p. 335.
[4] Anders M.N. Niklasson,
C.J. Tumczak, Matt Challacombe.
Trace resetting density matrix purification in O(N)
self-consistent-field theory J. of Chemical Physics, v. 118 N 15, 2003, pp.
1-10.
[5] А.М. Чернецов, О.Ю. Шамаева
“ Параллельная реализация метода Пальцера-Манолополиса для вычисления матрицы
плотности в задачах расчета электронной структуры молекул ”, в сб.
“Международный форум информатизации МФИ-2005. Труды международной конференции “Информационные средства и технологии”,
том 2, Москва,18-20 октября
[6] С.
Писсанецки, “Технология разреженных матриц”, М., Мир, 1988, 412 с.
[7] Дж. Голуб, Ч. Ван Лоун, “Матричные вычисления”, М., Мир, 1999,
550 с.
[8] SGI Computational Chemistry Applications
Performance Report. Spring 2002, Silicon Graphics, Inc.,
2002.